Die Partikelzählung in pharmazeutischen Anwendungen kann eindeutig in drei Kategorien eingeteilt werden: Zertifizierung, Qualifizierung und Überwachung. Jede Kategorie erfordert einen anderen Ansatz.
Zertifizierung: Messung eines Reinraums nach einer Norm. Die einzige weltweit anerkannte Norm ist ISO14644-1, "Klassifizierung der Luftreinheit anhand der Partikelkonzentration", die definiert, wie ein Reinraum funktioniert und ob er in der Lage ist, im gesamten Raum eine gleichmässige Luftqualität zu gewährleisten. Dies geschieht unabhängig von den darin ausgeführten Tätigkeiten.
Qualifizierung: Der Prozess der Analyse der Risikobewertung für die Tätigkeiten im Raum. Die Qualifizierung erfolgt nach den Prüfmethoden der Gittermethodik. Die Partikelzahl wird sowohl im Betrieb als auch im Ruhezustand gemessen; die Betriebsdaten sind jedoch am aussagekräftigsten.
Überwachung: Die fortlaufende Beprobung des Reinraums mit einer Häufigkeit, die dem Grad der Kontrolle entspricht, der erforderlich ist, um das Management des Risikos für das Endprodukt nachzuweisen. Die Anzahl der Probenahmestellen und ihr Standort werden durch die Risikobewertung und die Qualifizierungs- und Zertifizierungsprozesse bestimmt.
Zertifizierung
Wie bereits erwähnt, basiert die Zertifizierung von Reinräumen auf der Norm ISO14644-1, "Klassifizierung der Luftreinheit anhand der Partikelkonzentration". Die Einzelheiten der Bewertung können zwischen den FDA- und den EU-GMP-Vorschriften leicht variieren, aber die zugrunde liegende Methodik ist Standard. Mit der Zertifizierung wird nachgewiesen, dass der gesamte Bereich eine bestimmte ISO-Klassifizierung nach Partikelkonzentration erfüllt. Das heißt, unabhängig von der endgültigen Nutzung des Raums werden nur die Konzeption und die Ausführung des Filtersystems berücksichtigt. Die internationale Norm bedeutet, dass ein Reinraum, der auf die Einhaltung der ISO-5-Normen geprüft wurde, diese Norm unabhängig von geografischen und regulatorischen Aspekten (z. B. FDA oder EU-GMP) erfüllt. Damit steht ein universeller Standard zur Verfügung, der zeigt, dass ein Reinraumniveau erreicht wurde. Die Produkte von Particle Measuring Systems, darunter die Aerosolpartikelsensoren Airnet® II und IsoAir® Pro-E, entsprechen den neuen ISO-Normen von 2015. Die interaktive Software des Lasair® Pro Aerosolpartikelzählers kann den Benutzer sogar durch den Zertifizierungsprozess führen.
Es gibt viele verschiedene Mittel zum Nachweis der ISO-Konformität, auf die in diesem Beitrag nicht näher eingegangen werden soll. Am Beispiel einer klassischen Abfüllmaschine (Klasse A/ISO 5) in einem Bereich der Klasse B (ISO 7) können jedoch die Grundregeln der Prüfung aufgezeigt werden.
1. Die Anzahl der Probenahmestellen basiert auf einer statistischen Funktion der Fläche. Berechnen Sie die Fläche der Klasse A/ISO5. Bestimmen Sie die Anzahl der erforderlichen Probenahmestellen in der Tabelle.
- Berechnen Sie die Fläche des Grades A/ISO5. Bestimmen Sie die Anzahl der erforderlichen Probenahmestellen in der Tabelle.
- Berechnen Sie die Fläche des Grades B/ISO7. Ermitteln Sie die Anzahl der erforderlichen Probenahmestellen in der Tabelle.
2. Platzierung der Probenahmestellen für die Fläche des Grades A (ISO5):
- Die Probenahmestellen müssen alle in gleichem Abstand und auf Arbeitshöhe liegen, unabhängig von der Tätigkeit am Ort ihrer Platzierung.
- Die Proben werden in einem Raster an den festgelegten Stellen entnommen. Die Mindestanzahl der Probenahmestellen, NL, ist der ISO 14644-1 Tabelle A.1 zu entnehmen. Diese Tabelle gibt die Anzahl der Probenahmestellen in Bezug auf die zu klassifizierende Fläche eines jeden Reinraums oder einer reinen Zone an und bietet eine Sicherheit von mindestens 95%, dass mindestens 90% der Fläche des Reinraums oder der reinen Zone die Klassengrenzwerte nicht überschreiten.
- Die PASS/FAIL-Kriterien werden für ISO und EU-GMP Annex 1 berechnet. Es wird empfohlen, beide Datensätze zur Verfügung zu haben, da die FDA ISO14644-1 und die EU Annex 1 Datenpunkte verlangt (obwohl die EU-Daten für die FDA ausreichen würden).
3. Platzierung der Probenahmestellen für den Bereich der Klasse B (ISO7):
- Wiederholen Sie die Schritte, die für den Bereich der Klasse A (ISO5) verwendet wurden.
- Aufgrund der ungewöhnlichen Form eines Raums kann es schwieriger sein, die Standorte der Probenahmestellen zu bestimmen. Leiten Sie die Mindestanzahl der Probenahmestellen, NL, aus ISO 14644-1 Tabelle A.1 ab. Diese Tabelle gibt die Anzahl der Probenahmestellen in Bezug auf die Fläche jedes zu klassifizierenden Reinraums bzw. jeder zu klassifizierenden Reinzone an und bietet eine mindestens 95%ige Sicherheit, dass mindestens 90% der Fläche des Reinraums bzw. der Reinzone die Klassengrenzwerte nicht überschreiten.
4. Ein Abschlussbericht wird erstellt und markiert das Ende der Zertifizierungsphase.
Qualifizierung
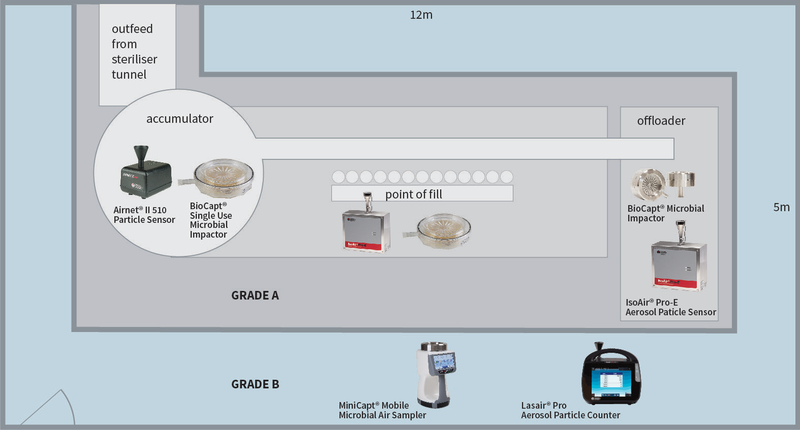
In der Qualifizierungsphase werden die Risiken für die Qualität des Endprodukts berücksichtigt. Jede Aktivität muss berücksichtigt und gemessen werden. Bleiben wir bei dem Beispiel der Abfüllanlage und betrachten wir den Akkumulatorentisch am Ausgang des Sterilisiertunnels. Das Risiko besteht darin, dass die Glaswaren (Fläschchen/Flaschen) der offenen Umgebung und dem Bediener ausgesetzt sind. Daher können Verunreinigungen in die sauberen Fläschchen/Flaschen vor der Abfüllung fallen. Das Eingreifen des Bedieners und das Verschieben der Glaswaren verursacht turbulente Luftbewegungen auf dem Tisch, die sich auf das Kontaminationsrisiko für die exponierten Fläschchen/Flaschen auswirken. Daher besteht in diesem Bereich ein Kontaminationsrisiko und es sollten die folgenden Massnahmen ergriffen werden:
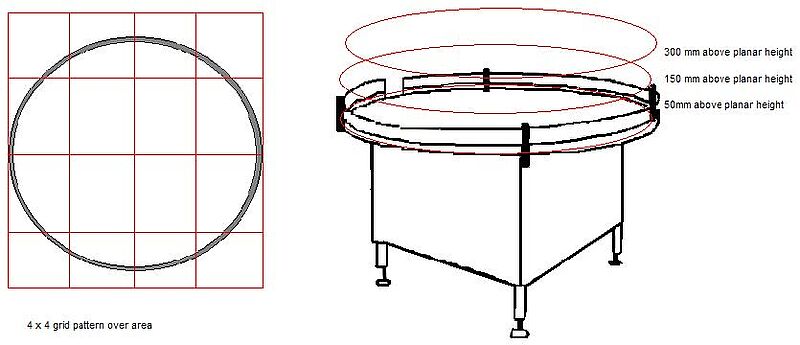
1. Teilen Sie den Risikobereich in ein 3 x 3 oder 4 x 4 Raster ein. Wenn die Tätigkeit auf mehreren Ebenen stattfinden kann, muss jede Ebene (Arbeitshöhe, +150 mm von der Arbeitshöhe und +300 mm von der Arbeitshöhe) berücksichtigt werden.
2. Entnehmen Sie eine Partikelprobe in der Mitte jedes Gitterquadrats auf jeder Ebene. - Die Proben werden in den Zuständen "In Ruhe" und "In Betrieb" entnommen. Es kann erforderlich sein, eine Tätigkeit oder einen Bediener zu umgehen, um geeignete Daten zu erhalten. - Geringfügige Verschiebungen der Probenahmestellen innerhalb des Rasterquadrats sind zulässig. Ein Standort ist ungültig, wenn er die normalen Aktivitäten behindert.
3. Wenn alle Proben genommen wurden, ergibt sich eine Partikelkarte der pharmazeutischen Aktivität. Jede der Hauptfunktionen innerhalb des Reinraums (Abfüllstelle, Verschließen, allgemeine Hintergrundaktivitäten usw.) sollte entsprechend analysiert werden.
Überwachung
Die Lage der Überwachungspunkte muss auf einer formalen Risikobewertung beruhen, bei der Instrumente wie die Fehler-Möglichkeits- und Einfluss-Analyse (FMEA) oder die Fehler-Möglichkeits-, Einfluss- und Kritikalitäts-Analyse (FMECA) mit Daten aus der Zertifizierungs- und Qualifizierungsprüfung verwendet werden, ohne darauf beschränkt zu sein. Andere Faktoren, wie z. B. Gerätestörungen, Montagepunkte, Bedienerimpedanz und Bedienereingriffe, tragen zur Auswahl der endgültigen Position für die Probensonde bei. Im derzeitigen regulatorischen Umfeld ist eine Risikobewertung unbedingt erforderlich. Ohne eine solche kann eine schlechte oder falsche Probenahmemethode dazu führen, dass Daten unzuverlässig mit dem Prozess in Verbindung gebracht werden. Dies könnte sich auch auf die Qualität des Endprodukts auswirken. Ohne die Möglichkeit, Ereignisse zu korrelieren, kann die fehlende Verbindung zwischen Standort und Probenahmehäufigkeit zu langwierigen Untersuchungen von Ereignissen führen, die außerhalb der Toleranz liegen. Die Festlegung eines risikobasierten Umweltüberwachungsplans erfolgt in mehreren Schritten:
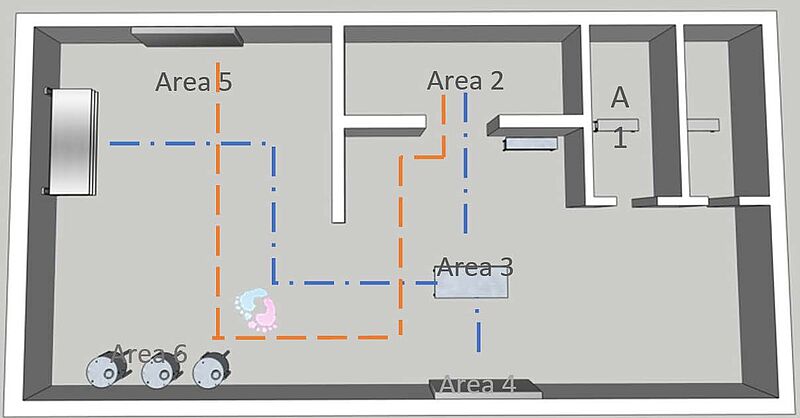
1. Prozessverständnis: Neben den Produktionsabläufen müssen auch die Personal- und Materialströme innerhalb des untersuchten Bereichs untersucht werden. Dadurch erhält man ein Verständnis dafür, wie das System genutzt wird und welche Anhaltspunkte es gibt, um den Kontrollzustand zu unterstützen, wie z. B.:
- Aktuelle Überwachungspraktiken
- Historische Daten
- Rauchstudien
Dieser Gemba-Spaziergang durch den Prozess und die Räume ist notwendig, um den Umfang der erforderlichen Überwachung zu definieren und die Anwendung eines Prozesses zu unterstützen, der zu den internen Praktiken einer Organisation passt. Die obige Abbildung ist ein Beispiel.
2. Definition der kritischen Bereiche: Mit Hilfe des HACCP-Konzepts (Hazard Analysis Critical Control Point) wird ermittelt, welche kritischen Bereiche eine Umgebungsüberwachung erfordern, und es werden die Bereiche identifiziert, die den Anforderungen eines kritischen Probenortes entsprechen.
3. Bewertung der Probenahmeverfahren: Sie müssen eine Entscheidung treffen zwischen traditionellen Methoden wie volumetrischen Luftprobennehmern, neueren Technologien wie mikrobiologischen Schnellmethoden oder manuellen Entnahmetechniken wie Abstrichen und Kontaktplatten. Bestimmen Sie auch, ob die gewählte Methode tragbar, kontinuierlich, ferngesteuert usw. sein muss.
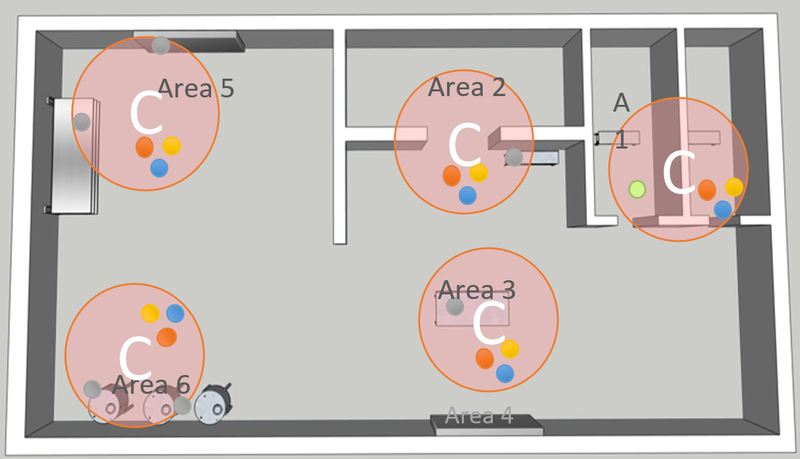
4. Festlegung der potenziellen Probenahmestellen: Bestimmen Sie einen einzelnen Probenahmeort innerhalb jedes kritischen Bereichs nach diesen Kriterien (wie in der nebenstehenden Abbildung dargestellt):
- Prüfen Sie den verfügbaren Platz um den kritischen Bereich.
- Messen Sie die Grösse von Sonden und Plattenhaltern.
- Bestimmen Sie die Zugänglichkeit des Ortes für die Wartung durch den Bediener.
- Beurteilen Sie die Wechselwirkung zwischen dem Prozessbetrieb und den Personal- und Materialströmen.
- Berechnen Sie die Wahrscheinlichkeit potenzieller Kontaminationsereignisse.
5. Definition der kritischen Kontrollpunkte (CCP): Jeder einzeln betrachtete Ort wird nach der FEMA-Methode bewertet, um eine Rangfolge zu erstellen und kritische Probenahmestellen zu ermitteln.
6. Definition der Probenahmeparameter: Die Häufigkeit der Probenahme wird auf der Grundlage der Kritikalität der Vorgänge sowie zusätzlicher Kriterien wie Inkubationsparameter und Abhilfemaßnahmen festgelegt, die vor der Erstellung eines Überwachungsplans eingeführt werden könnten. Zu den praktischen Aspekten der Probenahme gehören Elemente wie:
- Die isokinetische Probensonde sollte in den Luftstrom gerichtet sein.
- Die Mindestlänge der Schläuche sollte verwendet werden. Obwohl verschiedene Hersteller angeben, dass bestimmte Schlauchlängen mit ihrem Partikelzähler verwendet werden können, ist dies in der Regel eine Funktion der Dynamik der Vakuumpumpe und nicht des Partikeltransports. Partikel von 0,5 µm bewegen sich frei in langen Schläuchen. 5,0-µm-Partikel haben jedoch nicht dieselbe Beweglichkeit. Da 5,0-µm-Partikel ein grösseres Problem darstellen, sollten die Schläuche auf den kürzesten empfohlenen Längen gehalten werden1. Particle Measuring Systems gibt maximale Schlauchlängen an, die auf denselben Bedingungen des Luftstroms basieren, und empfiehlt eine maximale Länge von 3 m. Für pharmazeutische Partikelsysteme empfehlen wir jedoch eine reduzierte empfohlene Länge von 2 m, um den Transport der grösseren Partikel sicherzustellen.
Aus dem cGMP-Leitfaden der FDA für die aseptische Verarbeitung:
“Air in the immediate proximity of exposed sterilized containers/closures and filling/closing operations would be of appropriate particle quality when it has a per-cubic-meter particle count of no more than 3520 in a size range of 0.5 µm and larger when counted at representative locations normally not more than 1 foot away from the work site, within the airflow, and during filling/closing operations. This level of air cleanliness is also known as Class 100 (ISO 5).”
Mark Hallworth ist der Regionalmanager für Biowissenschaften bei Particle Measuring Systems. Er hat für pharmazeutische Gesellschaften in ganz Europa, Asien und den USA Vorträge über die Überwachung nicht lebensfähiger Partikel und Anlagen sowie über die Auswirkungen der Validierung dieser Systeme gehalten. Sie können ihn unter mhallworth(at)pmeasuring.com erreichen.